
When the Body’s Borders Break. Could Damage to Our Body’s Protective Surfaces Be Fueling the Rise of Chronic Disease?
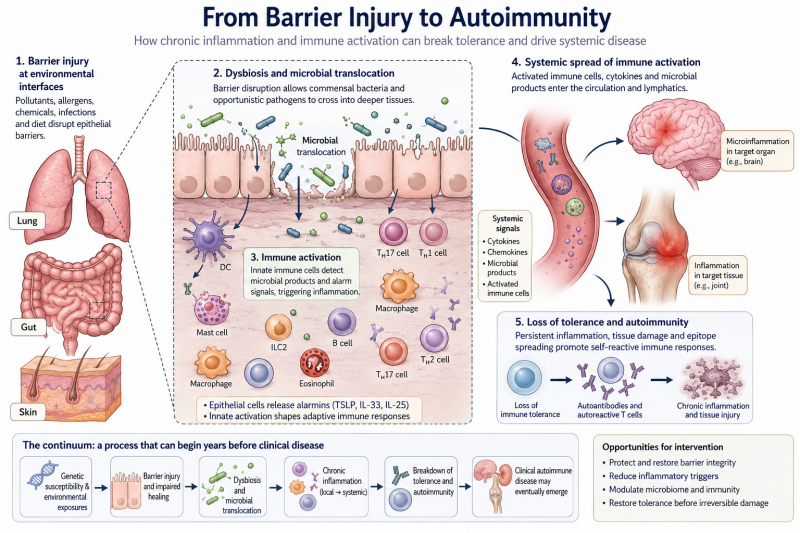
The path from a damaged body barrier to autoimmune disease may be a continuum, and understanding that progression could change when and how medicine intervenes
An autoimmune disease seems to begin on the day it is diagnosed. A young woman develops painful, swollen joints and is told she has rheumatoid arthritis. A child becomes desperately thirsty, loses weight and arrives at a hospital with type 1 diabetes. A patient with years of fatigue and strange neurologic symptoms finally undergoes an MRI that reveals the characteristic lesions of multiple sclerosis. The diagnosis feels like an event: a line dividing life into before and after.
But the immune system rarely changes that suddenly. Years before the first swollen joint, damaged pancreatic beta cell or neurologic lesion becomes clinically obvious, something may already be happening beneath the surface. Autoantibodies can appear. Immune-cell populations can shift. Microbial ecosystems can change. Epithelial barriers can become more permeable, while low-grade inflammation can spread through tissues without producing a recognizable disease.
By the time an autoimmune disease receives a name, the biological story may already be well underway. A growing body of immunology is therefore asking a different question. Instead of asking only what causes autoimmune disease, researchers are beginning to ask: How does immune tolerance gradually break?
The answer may connect several fields that medicine has traditionally studied separately: allergy, barrier biology, the microbiome, chronic inflammation and autoimmunity. The emerging picture is not of a single switch suddenly turning against the body. It is of a continuum. Barrier injury may create inflammation. Persistent inflammation may distort immune regulation. Repeated exposure to danger signals may expand self-reactive immune cells. Tissue damage may expose additional self-antigens. Eventually, an immune system that once tolerated the body may begin to attack it.
If that sequence is correct, the most important opportunity for preventing autoimmune disease may exist long before the disease has a name.
Tolerance Is an Active Achievement
The immune system faces a problem unlike almost any other system in biology. It must be capable of extraordinary violence while exercising extraordinary restraint. Every person carries T cells and B cells capable of recognizing an enormous range of molecular structures. This diversity allows the immune system to respond to viruses and bacteria it has never encountered before, but it also creates risk because some immune cells inevitably recognize molecules belonging to the body itself.
The immune system therefore maintains tolerance through multiple layers of control. During development, many strongly self-reactive immune cells are eliminated. Others are restrained in peripheral tissues. Regulatory T cells suppress inappropriate immune activation. Inhibitory receptors act as molecular brakes, while healthy tissues send signals encouraging immune calm.
Tolerance, then, is not simply the absence of immunity. It is an actively maintained state, and active systems can fail. Genetic susceptibility can weaken immune regulation. Infections can activate inflammatory pathways. Tissue injury can expose normally hidden antigens. Microbial molecules can stimulate innate immunity, and environmental exposures can repeatedly damage tissue.
Most of the time, these events do not cause autoimmune disease. But when several occur together, repeatedly and over time, tolerance may begin to erode.
The Barrier May Be Where the Story Starts
The skin, lungs and intestines are the largest points of contact between the immune system and the environment. They are also among the most immunologically complex tissues in the body. Healthy epithelial barriers maintain a carefully controlled relationship among environmental substances, resident microbes and immune cells. They allow enormous numbers of microorganisms to live close to the body without provoking continuous inflammation.
When a barrier is damaged, that relationship changes. Microbial products cross into deeper tissues. Allergens reach immune cells. Injured epithelial cells release alarmins and cytokines. Innate immune pathways become activated. If the injury heals, the system can return toward equilibrium. If it does not, inflammation may become persistent.
This is where the epithelial barrier hypothesis intersects with the biology of autoimmunity. Chronic barrier dysfunction may create an environment in which the immune system is repeatedly exposed to signals of injury, microbial invasion and tissue stress. The immune system is being asked, again and again, to decide whether something is dangerous. Under those conditions, the threshold for immune activation may change, and tolerance may become harder to maintain.
From Microinflammation to Systemic Inflammation
Inflammation is usually imagined as something obvious: a swollen ankle, a red rash or a fever. But inflammation can exist at scales too small to produce dramatic symptoms. At a damaged epithelial surface, small populations of immune cells may remain persistently activated. Cytokines may be produced locally. Microbial products may repeatedly enter deeper tissue, and the bone marrow may adjust the production and behavior of inflammatory cells.
Over time, what began locally may acquire systemic consequences. The bloodstream connects every tissue. Immune cells activated in one organ can circulate elsewhere. Cytokines influence distant tissues. Microbial molecules that cross a damaged intestinal or respiratory barrier can enter the circulation. The liver responds to inflammatory signals, and the bone marrow can change immune-cell production.
The concept of systemic inflammation does not necessarily mean that every organ is equally inflamed. It means that inflammatory information is no longer confined to its original location. The lung may influence the bone marrow. The intestine may influence the liver. The skin may shape systemic immune memory. A local barrier problem can become an organism-wide immunologic conversation.
The Immune System Learns From Context
One of the most important principles in immunology is that antigen recognition alone does not determine the immune response. Context matters. An immune cell encountering a protein in a calm environment may become tolerant to it. The same protein encountered alongside tissue damage, microbial products and inflammatory cytokines may provoke an immune response.
This principle helps explain why chronic inflammation is potentially dangerous for tolerance. The body contains countless self-antigens that the immune system normally ignores or actively tolerates. But damaged tissues release cellular proteins, DNA, RNA and other intracellular molecules into an inflammatory environment. The immune system now encounters self-antigens in the company of danger signals.
This does not automatically produce autoimmune disease. Multiple regulatory mechanisms still stand in the way. But repeated cycles of injury and inflammation may increase the probability that self-reactive immune cells escape control. Tolerance may not break in one dramatic moment; it may erode through repetition.
The Expanding Target
Once an autoimmune response begins, it may not remain focused on its original target. Tissue damage releases additional molecules. Those molecules can be captured by antigen-presenting cells and presented to T cells, allowing new immune responses to develop against additional components of the damaged tissue.
Immunologists call this epitope spreading. Imagine a fire beginning in one room of a house. The initial flame may be small and localized, but as surrounding structures burn, new materials become fuel. Autoimmunity can behave similarly. An immune response directed against one molecular target damages tissue. That injury exposes additional self-antigens. The immune response broadens, more tissue is injured and still more antigens are released.
A narrow immune abnormality can gradually become a complex disease. This may help explain why established autoimmune diseases are often much harder to reverse than early immune abnormalities. The disease is no longer driven by one signal. It has become an ecosystem.
Autoantibodies May Be Footprints, Not the Beginning
Modern medicine often discovers autoimmunity by detecting autoantibodies. Antinuclear antibodies, thyroid antibodies, rheumatoid factor, antibodies against citrullinated proteins and islet autoantibodies can reveal abnormal immune recognition. But an autoantibody does not always mean that clinical disease is present, and some autoantibodies appear years before symptoms.
This creates an important distinction between autoimmunity and autoimmune disease. Autoimmunity means that the immune system has developed measurable recognition of self. Autoimmune disease means that this process has progressed sufficiently to cause tissue dysfunction, organ injury or clinical symptoms.
Between these two states may lie an important window. A person can have immune abnormalities without irreversible organ damage, and this period may represent one of the most promising frontiers in preventive medicine.
The future of autoimmune care may increasingly resemble the prevention of cardiovascular disease. Medicine does not wait for a heart attack before measuring cholesterol or blood pressure. It identifies risk, tracks progression and intervenes before irreversible damage occurs. Autoimmunity may eventually be approached similarly.
Allergy and Autoimmunity May Be Less Separate Than They Appear
Allergy and autoimmunity have traditionally occupied different branches of immunology. Allergy was associated with IgE, mast cells, eosinophils and type 2 inflammation. Autoimmunity was associated with autoantibodies, autoreactive T cells and type 1 or type 17 inflammatory pathways. The distinction remains biologically useful, but the boundaries are becoming less rigid.
Both allergy and autoimmunity can involve damaged barriers, abnormal tissue signaling, dysregulated immune memory, altered microbiomes and failures of tolerance. Type 2 inflammation itself may interact with broader immune pathways. Epithelial alarmins can influence multiple immune-cell populations, while chronic tissue inflammation can alter antigen presentation and regulatory networks.
The question is therefore not whether allergy simply “turns into” autoimmune disease. That would be an oversimplification. The more interesting possibility is that both may emerge from overlapping disturbances in the relationship among tissue barriers, environmental exposures and immune regulation.
In one genetically susceptible person, the dominant expression may be asthma. In another, eczema and food allergy. In another, autoimmune thyroid disease. In some patients, these conditions coexist. The shared biology may lie upstream of the diagnostic labels.
The Lung May Be More Than a Breathing Organ
The lungs are particularly interesting in this emerging model. They possess an enormous epithelial surface exposed continuously to the external environment. Every day, thousands of liters of air pass through the respiratory tract, carrying particles, allergens, microorganisms, combustion products and chemical pollutants. Beneath this surface lies an extensive immune network.
For decades, asthma was largely conceptualized around narrowing of the airways. Modern immunology has expanded that view dramatically. The airway epithelium is now understood as an active immune sensor capable of releasing powerful signals such as TSLP, IL-33 and IL-25. These molecules can organize inflammatory responses far beyond simple bronchospasm.
Experimental work has also shown that the lung can influence the behavior of circulating immune cells. In animal models of neuroinflammatory disease, autoreactive T cells can pass through the lungs and undergo changes that affect their ability to enter the central nervous system. The lung, in other words, may function not only as a site of gas exchange but also as an immunologic training ground.
This raises an intriguing possibility: chronic airway inflammation may have consequences beyond respiratory symptoms. The full clinical importance of this idea remains uncertain, but it fits a broader principle increasingly recognized across immunology, immune events occurring at barrier organs can shape systemic immune behavior.
Why Disease Can Continue After the Trigger Is Gone
One of the most frustrating features of chronic inflammatory disease is persistence. A viral infection resolves, but symptoms continue. An environmental exposure ends, but inflammation remains. The original allergen is avoided, yet the tissue remains hyperresponsive.
One reason may be that biological systems can acquire memory. Adaptive immune cells form memory populations. Innate immune cells can undergo long-lasting functional reprogramming. Bone marrow progenitors can change their future inflammatory responses. Damaged tissues can undergo epigenetic changes that alter how they respond to later exposures.
The inflammatory system can become sensitized. A disease process may begin as exposure-dependent but later become partially self-sustaining. The first phase requires repeated injury; the later phase may require only small triggers to reactivate an already altered network. This is another reason early intervention matters. The longer inflammation persists, the more opportunities the immune system has to build memory around it.
Can Tolerance Be Restored?
For much of modern medicine, autoimmune disease has been considered controllable but not reversible. That assumption is beginning to be tested. Researchers are exploring antigen-specific immunotherapy, engineered regulatory T cells, tolerogenic dendritic cells, immune-resetting therapies and cell-based approaches designed not merely to suppress immunity but to restore tolerance.
The goal is fundamentally different from conventional immunosuppression. Instead of weakening the entire immune system, tolerance therapy seeks to teach the immune system which targets should no longer be attacked. Experimental studies in animals have shown that autoimmune disease can sometimes be reversed when regulatory immune networks are successfully restored, and early human studies in several autoimmune diseases are beginning to test related concepts.
But timing is likely to matter. Restoring immune tolerance cannot necessarily regenerate tissue that has already been permanently destroyed. A pancreatic beta cell lost to type 1 diabetes does not automatically return because the immune attack stops. Scarred tissue does not disappear simply because inflammation improves, and damaged neurons may have limited capacity for repair.
Future autoimmune medicine may therefore require two coordinated strategies: stop the immune attack and repair the organ. The earlier the first step occurs, the more tissue may remain available for the second.
The Continuum Before Disease
The emerging model of chronic inflammatory disease can be imagined as a progression: environmental exposure → barrier injury → epithelial alarm → dysbiosis → microbial translocation → local inflammation → systemic immune activation → loss of tolerance → autoimmunity → organ damage.
Reality is more complicated. Not every patient follows this sequence, different diseases have different genetic and environmental drivers, and some autoimmune diseases may begin through pathways unrelated to epithelial barrier failure. But the continuum is useful because it changes where medicine looks for opportunity.
Traditional care often begins near the end of the sequence. A patient develops symptoms, testing identifies organ dysfunction, a diagnosis is made and treatment begins. The emerging science asks whether the process can be recognized earlier. Can persistent barrier inflammation be identified? Can abnormal immune activation be measured before tissue injury? Can autoantibody trajectories reveal progression? Can environmental exposures be reduced while tolerance remains recoverable? Can inflamed tissues be repaired before the immune system builds durable memory against them?
These are no longer purely theoretical questions. They are becoming part of an emerging field of preventive immunology.
A Different Future for Chronic Disease
The greatest medical advances often occur when treatment moves earlier. Cardiology moved from treating heart attacks to controlling cholesterol and blood pressure. Oncology is moving from treating advanced cancer toward early detection and molecular interception. Infectious disease moved from treating AIDS-related complications to suppressing HIV before immune collapse.Immunology may be approaching a similar transition. The future may not be limited to controlling asthma attacks, suppressing eczema flares or slowing established autoimmune disease. It may involve identifying the biological progression that connects environmental injury to chronic inflammation and chronic inflammation to the loss of tolerance.
That progression may begin quietly: a damaged epithelial surface, a microbial community moving out of balance, a small amount of microbial translocation, a persistent cytokine signal, an immune cell that should have been restrained but remains activated, or an autoantibody appearing years before symptoms. None of these events alone necessarily causes disease, but together, over time, they may form a path.
The central challenge for medicine is to determine where along that path the process can still be redirected. Because the most consequential insight emerging from modern immunology may be that autoimmune disease does not begin when an organ fails. It begins earlier, during the long and largely invisible period when inflammation is changing the rules of immune tolerance.
If researchers can learn to recognize that period, the future of autoimmune medicine may be defined not only by suppressing disease after it appears, but by preventing the immune system from crossing the point of no return.
Reference
1. Akdis CA. Does the epithelial barrier hypothesis explain the increase in allergy, autoimmunity and other chronic conditions?. Nat Rev Immunol. 2021;21(11):739-751. doi:10.1038/s41577-021-00538-7
2. Odoardi F, Sie C, Streyl K, et al. T cells become licensed in the lung to enter the central nervous system. Nature. 2012;488(7413):675-679. doi:10.1038/nature11337
3. Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol. 2002;2(2):85-95. doi:10.1038/nri724
4. Deane KD, Holers VM. Rheumatoid Arthritis Pathogenesis, Prediction, and Prevention: An Emerging Paradigm Shift. Arthritis Rheumatol. 2021;73(2):181-193. doi:10.1002/art.41417
5. Herold KC, Bundy BN, Long SA, et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N Engl J Med. 2019;381(7):603-613. doi:10.1056/NEJMoa1902226